
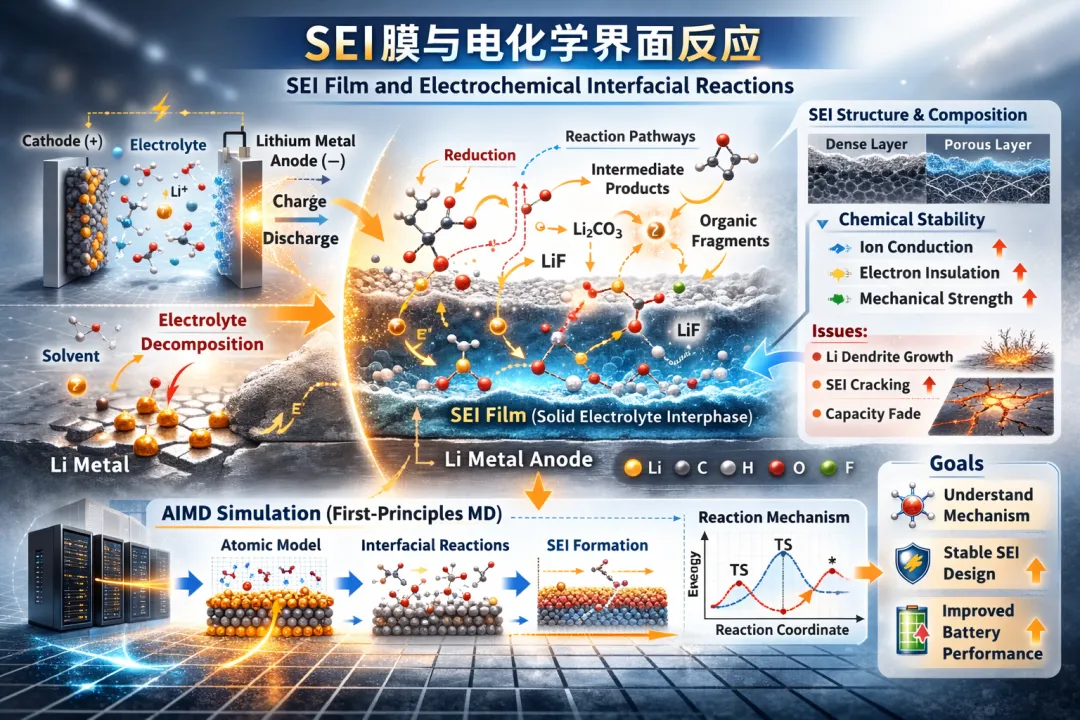
浪潮信息推动电池SEI膜研究突破:开源198原子量子化学模拟基准测试
随着新能源产业的快速发展,对高性能储能技术的需求日益迫切,无论是提升电动车的续航里程,还是保障智能电网的稳定运行,都离不开电池技术的突破。锂金属电池虽被誉为下一代储能技术的关键,但其大规模商业化进程却遭遇了阻碍,其中一个核心挑战在于金属电极与电解质分子界面的复杂化学反应。这一反应会生成固态电解质界面膜(SEI),该膜的稳定性直接决定了电池的循环寿命和安全性。然而,由于SEI膜的成分与结构极其复杂,且容易受到外部环境(如空气)的影响,它一直是锂电池研究中最关键也最难以深入解析的领域之一。因此,揭示SEI膜的形成机制,对于优化电池性能、推动新一代电池技术的研发,具有至关重要的意义。
想要深入研究这个问题,科学家们需要用到基于第一性原理分子动力学(Ab Initio Molecular Dynamics,AIMD)模拟,该方法可在原子尺度上描述化学反应过程。然而,模拟真实电池界面常需构建包含金属电极和溶剂离子的大型模型(通常达数百原子),并进行长时间尺度的动力学模拟,对计算平台的浮点性能、内存带宽与并行扩展能力提出极高要求。
近年来,处理器架构演进与计算技术的发展,显著提升了单节点能力与并行规模。然而,硬件性能的发挥依赖于具有代表性的基准算例,以全面评估计算软件在实际科研场景中的并行效率、内存访问模式以及I/O性能。当前公开的材料模拟测试算例大多规模偏小,往往只包含几十个原子,无法还原真实反应路径,也就无法暴露大规模并行计算中的通信瓶颈、内存带宽压力等关键问题。因此,构建贴近真实科研需求的大规模模拟算例,对评估计算平台在材料科学领域的适用性至关重要。
为此,浪潮信息研究团队基于Nature Chemistry前沿研究工作,构建了一个利用开源第一性原理计算软件Quantum ESPRESSO、面向电池界面反应研究的AIMD测试算例。该算例模拟了铜–锂(Cu–Li)金属界面与电解质分子之间的反应过程,具有以下几个显著特点:
首先,体系规模大,计算负载高。体系包含198个原子、7种化学元素,完整模拟了金属界面与电解质分子的复杂交互,对处理器浮点性能和内存带宽提出了较高要求,能够有效体现现代服务器平台的计算能力。
其次,并行扩展性良好。Quantum ESPRESSO具有良好的并行特性,该算例能够在多节点环境中实现高效扩展,可用于评估服务器平台在第一性原理计算中的并行性能。
此外,科学价值突出。AIMD模拟能够从原子尺度揭示界面反应机理,对于理解SEI形成过程和提升电池稳定性具有重要意义。因此,该算例对于当前的科学研究计算需求来说,具有广泛的代表性。
该算例兼具科研价值与计算复杂度,既可服务于电池界面反应的研究,也为评估现代服务器系统在第一性原理分子动力学模拟中的性能表现提供了可靠基准。此算例已开源至github平台(https://github.com/hpc-testkit/QE-benchmark-SEI)。
1 Quantum ESPRESSO:兼具精度与复杂度的第一性原理工具
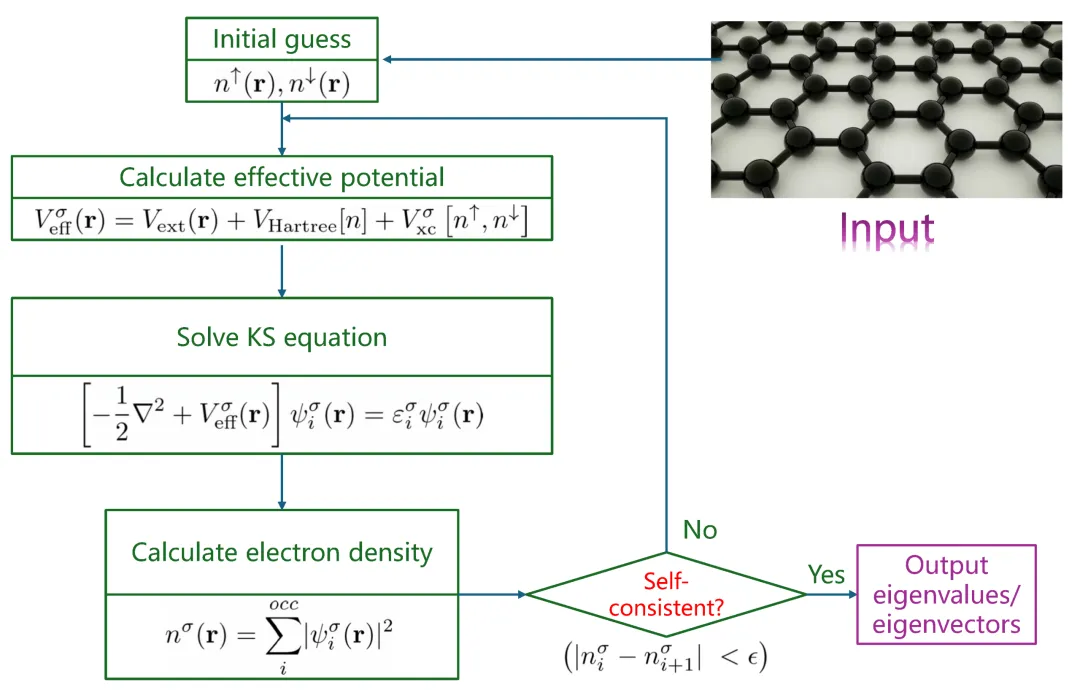
第一性原理电子结构计算是现代计算材料科学中最重要的工具之一。自20世纪90年代以来,随着密度泛函理论(Density Functional Theory, DFT)的发展及计算技术进步,第一性原理方法已广泛应用于材料结构预测、电子性质分析及化学反应机理研究等领域。
Quantum ESPRESSO是国际上广泛使用的开源第一性原理软件,由意大利里雅斯特CNR-IOM DEMOCRITOS国家仿真中心发起,并与全球多所研究机构合作,如MIT、Princeton University、University of Minnesota及EPFL。软件基于平面波基组和赝势方法实现DFT计算,并通过多层并行策略(k点、能带和FFT并行)支持大规模体系模拟。
与原子轨道基组方法相比,平面波基组精度可控、系统性好,适合周期性固体体系。Quantum ESPRESSO提供基于第一性原理的Born–Oppenheimer分子动力学(BOMD)和Car–Parrinello分子动力学(CPMD),可直接模拟化学键断裂与界面反应等动力学过程。由于每个时间步都需完成电子自洽计算,其计算量远高于传统分子动力学,当体系超过上百原子时,计算复杂度显著增加,因此该软件也是高性能服务器浮点计算能力与并行效率测试的重要工具。
2 基准算例:198个原子组成的铜-锂界面反应
本基准算例基于第一性原理分子动力学(AIMD),用于模拟Cu–Li金属界面与电解质FSI⁻离子之间的反应过程。体系由Cu金属基底、界面Li层、惰性He隔离层以及由S、N、O、F构成的电解质分子组成,共包含198个原子、7种元素。通过在界面法向(x方向)引入80Å的大晶胞长度(整体尺寸为80×11×11ų),有效降低周期性边界条件带来的相互作用。电子结构计算采用基于PBE泛函的密度泛函理论(DFT),平面波截断能分别设置为37Ry(波函数)和368Ry(电荷密度),并结合PAW与ultrasoft赝势,在保证精度的同时控制计算成本;针对金属体系,采用Methfessel–Paxton展宽(0.1eV)提升自洽收敛稳定性,k点采样使用1×2×2网格以平衡精度与效率。动力学模拟中采用Verlet积分算法,时间步长为20a.u.,示例运行5步(实际应用通常需数千步以上),并通过温度重标定方法将体系控制在300K,近似描述有限温度下的界面反应行为。
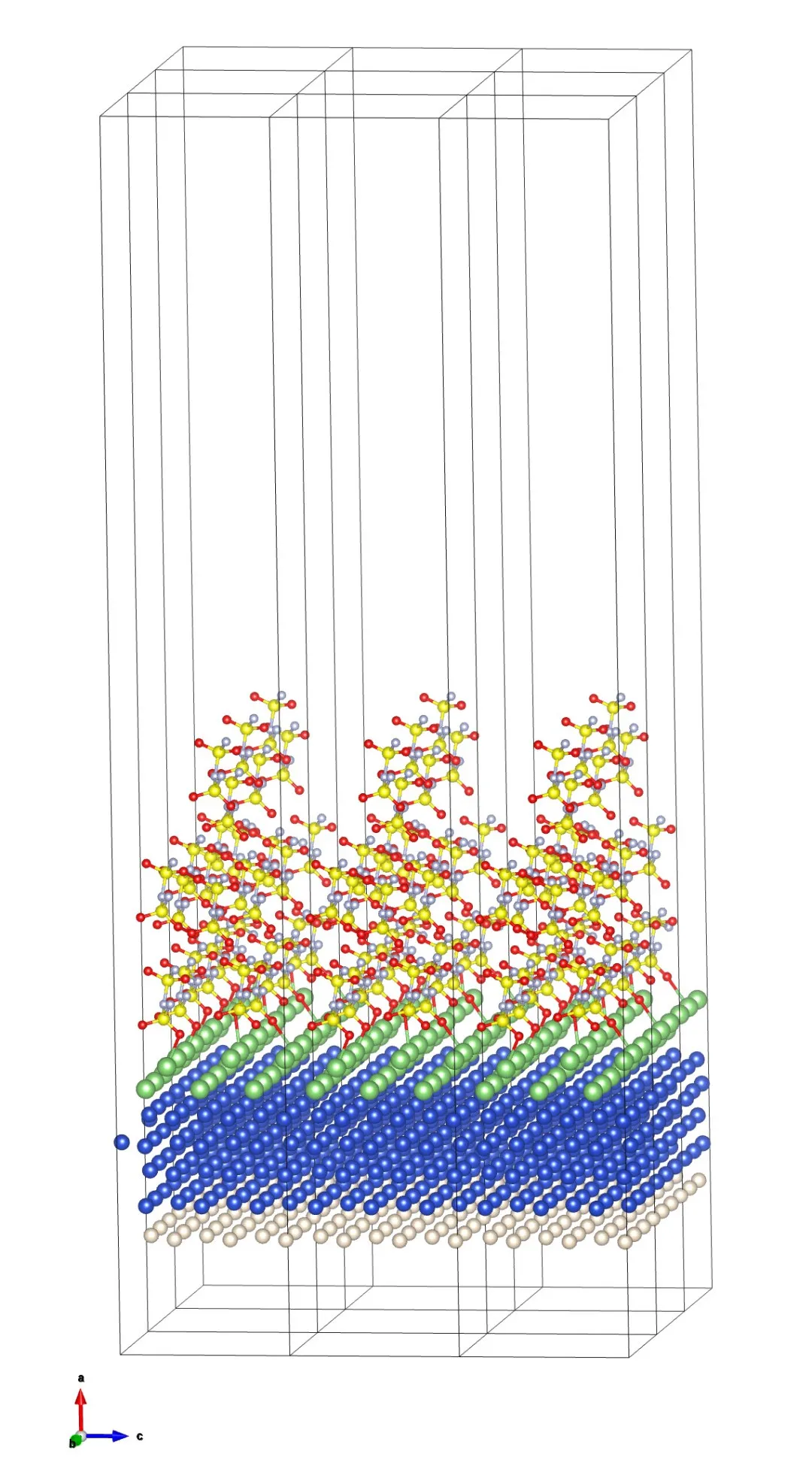
图3 Cu–Li金属界面与电解质分子之间的分子动力学模拟结构图(1×3×3超胞结构)。其中由下至上有三层,分别为白色球(He原子层)、蓝色球(Cu原子层)和绿色球(Li原子层);上面的FSI-离子包含黄色球(S原子)、红色球(O原子)、连接两个S原子的银色球(N原子)和只连接一个S原子的银色球(F原子)。
3 模拟结果:一套能评估服务器真实性能的测试用例
3.1 模拟设置:还原真实科研条件
在Quantum ESPRESSO AIMD模拟前,我们首先对构建的Cu–Li界面体系和电解质分子分别进行了结构优化,以避免初始构型中原子位置不合理。在此基础上构建了此算例用于开展基于Born–Oppenheimer近似的第一性原理分子动力学(AIMD)模拟。整个过程在接近室温的恒温条件下进行,通过温度控制算法维持体系热平衡,体系在极短时间尺度(飞秒级)下进行了AIMD演化,以捕捉界面反应过程及其结构与能量变化。在每一个时间步中,均需完成电子结构自洽计算、原子受力评估及原子位置更新等关键步骤;由于体系规模较大且涉及复杂界面电子结构,整体计算具有较高的计算负载与性能挑战。
3.2 模拟过程:短时间也有高负载
本算例通过短时间(nstep=5)的第一性原理分子动力学模拟进行试探性模拟,以控制计算成本并验证模拟流程,展示了电解质分子在Cu–Li界面附近的初步热运动与界面相互作用。尽管原子位置变化有限,尚未形成完整的化学键重构或固态电解质界面(SEI),模拟过程已完整涵盖电子结构自洽计算、原子受力评估及结构更新等关键步骤,真实反映了大规模界面体系的动力学演化特征。由于体系包含198个原子、多元素混合及复杂界面电子结构,即便在短时间尺度下计算负载亦极高,对平台的计算能力、内存管理及并行效率提出了严格挑战。因此,该算例不仅为界面化学动力学提供了初步物理洞察,同时也可作为计算平台性能和并行策略评估的基准案例。
3.3 算例优势:比常见示例更具代表性
与常见的Quantum ESPRESSO示例算例相比,本研究构建的AIMD算例在体系规模、结构复杂度以及研究问题方面均更具代表性。该体系包含约200个原子和多种元素(Cu、Li、S、N、O、F等),用于模拟锂金属电池中电解质分子在金属界面上的分解反应以及固态电解质界面(SEI)的形成过程。这一界面反应机制是当前高能量密度电池研究中的前沿热点问题,对于理解电池稳定性和界面调控具有重要意义。与传统的小规模晶体结构测试算例相比,该算例采用第一性原理分子动力学(AIMD)模拟,需要在每一个时间步执行完整的电子结构自洽计算,同时复杂的多元素界面体系也显著增加了电子结构计算规模,使其能够产生更高的计算负载。因此,该算例不仅具有较高的科研应用价值,同时也能够更加真实地反映第一性原理材料模拟中的计算特征,适合作为评估高性能服务器平台在大规模材料计算场景下性能表现的测试用例。
4 总结
本研究成功构建了一套基于Quantum ESPRESSO软件的第一性原理分子动力学(AIMD)基准测试案例,专门用于模拟铜-锂(Cu-Li)金属界面与电解质分子之间的复杂交互反应。该测试案例包含198个原子,涉及7种不同的化学元素,体系结构复杂且计算负载显著,能够真实模拟材料科学实际研究中服务器所承受的高强度计算压力。在确保第一性原理计算物理精确度的基础上,该案例可用于系统性地评测服务器平台在多个核心计算环节的性能表现,具体包括:处理电子结构计算时的浮点运算能力、执行大规模AIMD模拟时的并行扩展效率,以及对内存带宽和I/O子系统的高效访问能力。